This web page was produced as an assignment for Genetics 564, an undergraduate course at UW-Madison.
conclusions
Alzheimer's disease, though linked to several genes, is a direct causation of several deletions in the PSEN1 gene [1]. Because phenotypic symptoms such as memory loss, disorientation and incapacity for everyday tasks do not have an early onset, Alzheimer's can be both difficult to diagnose and treat. When PSEN1 is mutated, the body produces APP at an increasingly abnormal rate, which is then degraded into various neurotoxic subcomponents via gamma-secretase [2] [3]. These subcomponents then cause the formation of amyloid plaques and neurofibrillary tangles, which lead to the affected brain and neural tissue observed during Alzheimer's.
As PSEN1 is a relatively conserved gene across various organisms, there are effectively many models to study Alzheimer's with. The conservation of PSEN1 also yields high conservation of PSEN1, the protein for which PSEN1 codes for. Being a regulatory protein, PSEN1 is known to interact with many other proteins of varying functions. Through the STRING database, PSEN1 is linked to other proteins that function in cell adhesion, cellular transport, cell signaling, and the gamma-secretase subunit; however, STRING also highlights the interaction between PSEN1 and UBQLN1, a ubiquitin protein correlated to PSEN1 over-accumulation [4].
As PSEN1 over-accumulation would have a direct causation on the formation of amyloid plaques, the role of UBQLN1 in Alzheimer's can be inferred. Despite this connection, it is currently unknown if the downregulation of UBQLN1 could mediate the plaque formation observed in Alzheimer's. Using various genomic, proteomic, and bioinformatic tools, I hypothesize that UBQLN1-mediated ubiquitination is required to regulate PSEN1 activity in regards to APP production.
As PSEN1 is a relatively conserved gene across various organisms, there are effectively many models to study Alzheimer's with. The conservation of PSEN1 also yields high conservation of PSEN1, the protein for which PSEN1 codes for. Being a regulatory protein, PSEN1 is known to interact with many other proteins of varying functions. Through the STRING database, PSEN1 is linked to other proteins that function in cell adhesion, cellular transport, cell signaling, and the gamma-secretase subunit; however, STRING also highlights the interaction between PSEN1 and UBQLN1, a ubiquitin protein correlated to PSEN1 over-accumulation [4].
As PSEN1 over-accumulation would have a direct causation on the formation of amyloid plaques, the role of UBQLN1 in Alzheimer's can be inferred. Despite this connection, it is currently unknown if the downregulation of UBQLN1 could mediate the plaque formation observed in Alzheimer's. Using various genomic, proteomic, and bioinformatic tools, I hypothesize that UBQLN1-mediated ubiquitination is required to regulate PSEN1 activity in regards to APP production.
aim 1
The purpose of the first aim was to identify a drug compound that could be used to inhibit UBQLN1 function of PSEN1 accumulation. If PSEN1 accumulation could be regulated in those with Alzheimer's, the formation of amyloid plaques could be controlled, slowing the disease progression. Due to the accessibility, I propose the use of model zebrafish to establish a direct target library.
Using PubChem and using the bioassay search function in regards to Alzheimer's yielded 9,567 plausible drug compound results. Though many of these compounds have yet to be tested and confirmed, several have been established to affect PSEN1 function. As such, I deduce that some of these predicted compounds may be capable of binding to and regulating UBQLN1 function as well.
Using PubChem and using the bioassay search function in regards to Alzheimer's yielded 9,567 plausible drug compound results. Though many of these compounds have yet to be tested and confirmed, several have been established to affect PSEN1 function. As such, I deduce that some of these predicted compounds may be capable of binding to and regulating UBQLN1 function as well.
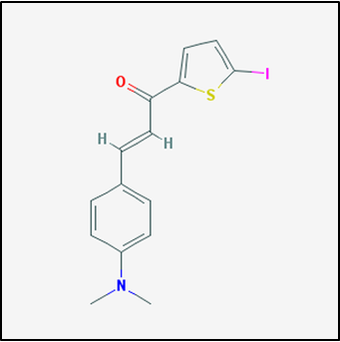
Figure 1. Drug compound 17757413
|
To find a plausible treatment, a direct target library will be established using mutant zebrafish and a 96-well plate. This technique will yield at least one compound that will bind to UBQLN4, the homolog to UBQLN1 in zebrafish.
Observed to the left is compound 17757413, one of the compounds known to affect PSEN1. Using this drug, I hypothesize that treating mutant zebrafish will supress the formation of APP and amyloid plaques. From here, a memory test can be performed on the model zebrafish, specifically using a T-test designed for zebrafish. By observing the neural tissue of the fish, the amount of amyloid plaques formed can be determined. |
As mentioned, I would expect a zebrafish with mutated UBQLN4 and treatment with a compound like 17757413 to have suppressed plaque formation, as well as a positive memory test result. As such, this aim will conclude that drug compounds capable of regulating ubiqutination will be a direct causation on the number of plaques observed.
aim 2
The second aim will be used to identify additional proteins that may regulate PSEN1 during the Alzheimer's disease state. As such, I hypothesize that other ubiquitination-inducing proteins will be linked to the formation of amyloid plaques observed in the disease phenotype. To proceed with this aim, I propose the use of tandem affinity purification, or a TAP-tag method in model mice, due to the higher correlation of their PSEN1 to that of humans.
By TAP-tagging UBQLN1 in mice, brain-specific candidate proteins can be identified. Though there may be several results, a western blot could be used to observe the binding affinity of the these novel proteins to UBQLN1. Once a binding protein is chosen, for example Psmd4 in mice, CRISPR can then be used to for a complete knockout.
By TAP-tagging UBQLN1 in mice, brain-specific candidate proteins can be identified. Though there may be several results, a western blot could be used to observe the binding affinity of the these novel proteins to UBQLN1. Once a binding protein is chosen, for example Psmd4 in mice, CRISPR can then be used to for a complete knockout.
|
Seen to the right is mouse brain, one with amyloid plaques and one without--left to right, respectively. It is my hypothesis that a knockout of the identified binding proteins will cause the formation of amyloid plaques, even in healthy, wild type PSEN1 mice.
Using a memory test to correlate the results, I expect these novel binding proteins to be found essential for regulating PSEN1 and UBQLN1 function, and that a CRISPR knockout will cause Alzheimer's phenotypic symptoms to develop. Thus, new proteins linked to Alzheimer's can be identified and further studied in the future. |
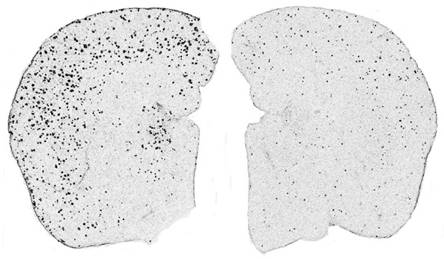
Figure 2. Amyloid plaques observed in mice brains
|
aim 3
The last aim is targeted to determine the importance of certain post-translational modifications made in PSEN1. This would be done using NetPhos 2.0, as well as Clustal Omega to determine amino acid site-specific conservation across organisms. Though many organisms were used to identify conserved post-translational modifications, the ones included below are humans and the two model organisms, mice and zebrafish.
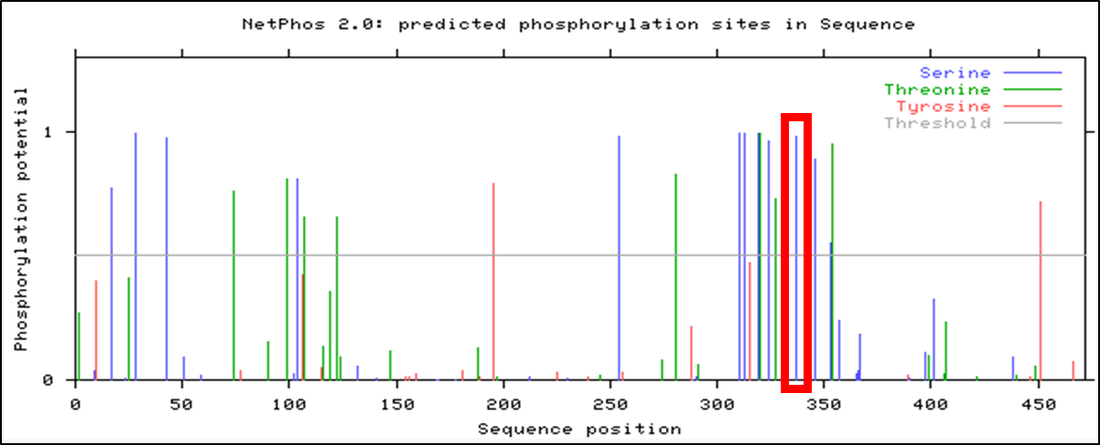
Figure 3. NetPhos 2.0 result for Human PSEN1
Observed above is the NetPhos 2.0 result for human PSEN1. Though several phosphorylation sites were identified, only a few fell within the active presenilin domain of the PSEN1 protein, narrowing the selection for the most conserved post-translational modification. In addition, this NetPhos 2.0 result was then compared between mice and zebrafish, observed below.
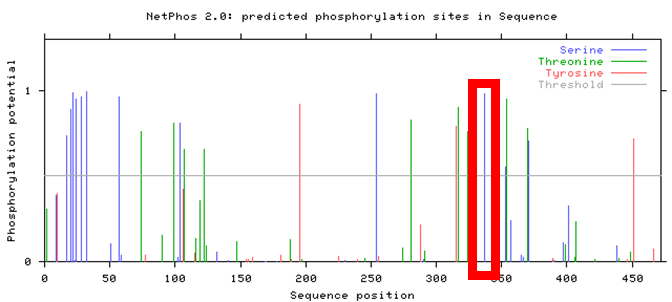
Figure 4. NetPhos 2.0 result for Mice PSEN1
|
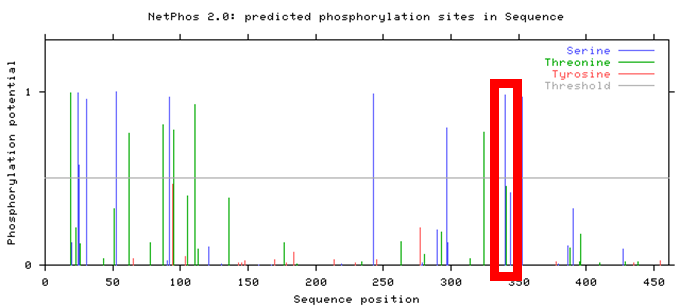
Figure 5. NetPhos 2.0 result for Zebrafish PSEN1
|
Comparing these three results with Clustal Omega seen here identify areas of high conservation of PSEN1 amino acid sites across various organisms. Though several phosphorylation sites reside within the active presenilin domain, the results illustrate the highest conservation for serine-320 in human PSEN1, highlighted in each of the above NetPhos 2.0 results.
After identifying serine-320, I propose the use of CRISPR to alter this amino acid from serine to arginine. In doing so, the amino acid will not be phosphorylated, thus altering the protein function. Because of this alteration, I expect amyloid plaques to form in the individual, causing a failed memory test and the progression of Alzheimer's disease symptoms. Likewise, the chosen serine could be replaced with aspartic acid, causing the protein to believe it is continuously phosphorylated, which should yield results similar to that of arginine, altering PSEN1 function.
This information then signifies the importance of certain post-translational modifications in PSEN1, specifically serine-320 in humans. As such, this can then be a large cornerstone of future research in regards to what is a direct causation of Alzheimer's disease and the related phenotypic symptoms.
After identifying serine-320, I propose the use of CRISPR to alter this amino acid from serine to arginine. In doing so, the amino acid will not be phosphorylated, thus altering the protein function. Because of this alteration, I expect amyloid plaques to form in the individual, causing a failed memory test and the progression of Alzheimer's disease symptoms. Likewise, the chosen serine could be replaced with aspartic acid, causing the protein to believe it is continuously phosphorylated, which should yield results similar to that of arginine, altering PSEN1 function.
This information then signifies the importance of certain post-translational modifications in PSEN1, specifically serine-320 in humans. As such, this can then be a large cornerstone of future research in regards to what is a direct causation of Alzheimer's disease and the related phenotypic symptoms.
future directions
Going forward, we must narrow the complexity of Alzheimer's disease. Though several genes and proteins are involved in causing the aforementioned disease state, focusing on PSEN1 is a decent place to start. By centering modern research on PSEN1 and UBQLN1, we may be able to target a more direct causation for Alzheimer's disease.
In addition, the above aims address where future research should lie: UBQLN1, various binding proteins and the highly conserved post-translational modifications like serine-320. Together, this research could identify plausible treatments for the phenotypic symptoms, whether it be through drug compounds or methods such as CRISPR. This will also provide insight to the importance of UBQLN1 and ubiquitination on various neurological diseases.
If this research is taken into consideration, we may then be one step closer to curing the worldwide neurological disease known as Alzheimer's.
In addition, the above aims address where future research should lie: UBQLN1, various binding proteins and the highly conserved post-translational modifications like serine-320. Together, this research could identify plausible treatments for the phenotypic symptoms, whether it be through drug compounds or methods such as CRISPR. This will also provide insight to the importance of UBQLN1 and ubiquitination on various neurological diseases.
If this research is taken into consideration, we may then be one step closer to curing the worldwide neurological disease known as Alzheimer's.
| cortezpresentation_finaldraft.pdf |
| cortezpresentation_finaldraft.pptx |
| cortezpresentation_roughdraft2.pptx |
| cortezpresentation_roughdraft.pptx |
REFERENCES
Cover picture: obtained from http://exelmagazine.org/article/an-alzheimers-cure-for-fruit-flies/
[1] De Strooper, B. (2007). Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease.EMBO Reports, 8(2), 141–146. doi:10.1038/sj.embor.7400897
[2] Selkoe D (1994). "Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer's disease". Annu. Rev. Cell Biol. 10: 373-403.doi:10.1146/annurev.cb.10.110194.002105.PMID 7888181.
[3] Barnwell, E., Padmaraju, V., Baranello, R., Pacheco-Quinto, J., Crosson, C., Ablonczy, Z., … Sambamurti, K. (2014). Evidence of a Novel Mechanism for Partial γ-Secretase Inhibition Induced Paradoxical Increase in Secreted Amyloid β Protein. PLoS ONE, 9(3), e91531. doi:10.1371/journal.pone.0091531
[4] Mah, Alex L., George Perry, Mark A. Smith, and Mervyn J. Monteiro. "Identification of Ubiquilin, a Novel Presenilin Interactor That Increases Presenilin Protein Accumulation." National Center for Biotechnology Information. N.p., 13 Nov. 2000. Web. 12 Apr. 2015. <http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2169435/pdf/0002132.pdf>.
[1] De Strooper, B. (2007). Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease.EMBO Reports, 8(2), 141–146. doi:10.1038/sj.embor.7400897
[2] Selkoe D (1994). "Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer's disease". Annu. Rev. Cell Biol. 10: 373-403.doi:10.1146/annurev.cb.10.110194.002105.PMID 7888181.
[3] Barnwell, E., Padmaraju, V., Baranello, R., Pacheco-Quinto, J., Crosson, C., Ablonczy, Z., … Sambamurti, K. (2014). Evidence of a Novel Mechanism for Partial γ-Secretase Inhibition Induced Paradoxical Increase in Secreted Amyloid β Protein. PLoS ONE, 9(3), e91531. doi:10.1371/journal.pone.0091531
[4] Mah, Alex L., George Perry, Mark A. Smith, and Mervyn J. Monteiro. "Identification of Ubiquilin, a Novel Presenilin Interactor That Increases Presenilin Protein Accumulation." National Center for Biotechnology Information. N.p., 13 Nov. 2000. Web. 12 Apr. 2015. <http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2169435/pdf/0002132.pdf>.
